| |
Light Microscopy: Principles & Applications
Live Cell Microscopy and Time Lapse Analysis
Principle:
In Live Cell Microscopy, not only imaging quality is an important factor, but the cells have to be kept in a healthy condition. Fluorophores as well as light exposure can alter the behaviour of the cells significantly. Thus, cells should be kept at their favoured temperature (e.g. 37 °C) and light exposure should be minimized. With decreasing wavelength, photodamage is increased. Use of genetically encoded fluorescent proteins allows direct observation of proteins in their native environment. Changes in the localization and binding of fluorescent proteins over time can be easily accessed. For time lapse analysis,
alignment of the structures over time is crucial and can be a difficult task, as cells do not only move but can also change their shape over time.
References:
Easwaran, H.P., Leonhardt, H., Cardoso, M.C. (2004)
Martin, R.M.; Leonhardt, H., Cardoso M.C. (2005)
Rothbauer, U., Zolghadr, K., Tillib, S., Nowak, D., Schermelleh, L., Gahl, A., Backmann, N., Conrath, K., Muyldermans, S., Cardoso, M. C., and Leonhardt, H. (2006)
Further Readings:
http://micro.magnet.fsu.edu/primer/techniques/livecellimaging/index.html
http://zeiss-campus.magnet.fsu.edu/articles/livecellimaging/index.html

|
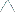 |
Fluorescence Photobleaching
Principle:
In photobleaching techniques like Fluorescence Recovery after Photobleaching (FRAP) or Fluorescence Loss In Photobleaching (FLIP) allow studying interaction times at determined sites within the cell. In FRAP, a region of interest is bleached and the speed and completeness of the fluorescence recovery measured, while in FLIP a region of interest is repeatedly photobleached and the fluorescence loss in other parts of the cell is monitored. Depending on the acquisition rate and the system of interest, diffusion or binding and release at certain positions in the observed area can be the
dominant features for the fluorescence recovery (FRAP) or loss (FLIP). |
FRAP |
 |
FLIP |
 |
| |
References:
Sporbert, A., Gahl, A., Ankerhold, R., Leonhardt, H., and Cardoso, M.C. (2002)
Sporbert, A., Domaing, P., Leonhardt, H. and Cardoso, M. C. (2005)
Schermelleh, L., Haemmer, paA., Sda, F., Rosing, N., Meilinger, D., Rothbauer, U., Cardoso, M. C., and Leonhardt, H. (2007)
Lengert, L., Lengert, N., Drossel, B., Cardoso, M. C., Muster, B., Nowak, D. and Rapp, A. (2015)
Further Readings:
http://micro.magnet.fsu.edu/primer/java/fluorescence/photobleaching/index.html
https://www.olympus-lifescience.com/en/microscope-resource/primer/techniques/confocal/applications/flipandfrap/
|
|
Fluorescence Photoactivation / Conversion
Principle:
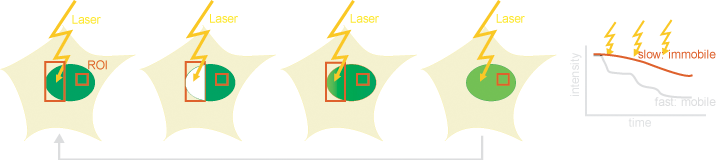
Photoactivable mutants of the green fluorescent protein (GFP) have opened new ways of investigating binding and diffusion kinetics in living cells. These mutants have a low absorbance at 488nm, were GFP is normally excited. Upon radiation with 400nm, a decarboxylation in the GFP is triggered which is accompanied with a conformational change resulting in an increase of the absorbance at ~488nm, and in the microscopy image, the dye gives a bright stable fluorescence. This way, a determined location within a cell can be activated and diffusion and binding kinetics can be locally measured. Thereby mainly off-kinetics are obtained.
Additionally, photoconvertible proteins have been reported, which can switch their spectral properties upon irradiation (e.g. Dendra).
|
 |
References:
Schermelleh, L., Spada, F., Easwaran, H.P., Zolghadr, K., Margot, J.B., Cardoso, M.C.and
Leonhardt, H. (2005)
Further Readings:
https://www.olympus-lifescience.com/en/microscope-resource/primer/techniques/confocal/applications/opticalhighlighters/
https://www.microscopyu.com/techniques/fluorescence/introduction-to-fluorescent-proteins
|
|
Fluorescence Correlation Spectroscopy
Principle:
In Fluorescence Correlation Spectroscopy (FCS), fluorescence changes within a defined volume, in general determined by the confocal detection volume, are monitored with a very high time resolution. Use of single molecule
sensitive avalanche diodes as detectors allows monitoring intensity changes due to entrance of leaving of single molecules. By mathematical conversion of the fluorescence intensity trace into the so-called autocorrelation function, information about sample concentration and diffusion coefficients can be obtained. Also binding kinetics or photophysical processes can be analyzed.
References:
Grünwald, D., Cardoso, M. C., Leonhardt, H., Buschmann, V. (2005)
Further Readings:
http://zeiss-campus.magnet.fsu.edu/articles/livecellimaging/techniques.html
|
|
Förster (Fluorescence) Resonance Energy Transfer & Fluorescence Lifetime Imaging Microscopy
Principle:
Fluorescence Resonance Energy Transfer (FRET) is the nonradiative transition of the excitation energy of an excited donor dye to an acceptor dye. The efficiency of the transfer depends on the distance between the donor dyes, their relative orientation and the spectral properties. For good FRET-pairs that show a big overlap between the donor emission and the acceptor absorption maximum, FRET is observed for distances
up to 10 nm. The resonance energy transfer quenches donor emission, resulting in a reduction of fluorescence intensity and lifetime, while the acceptor starts emitting. In cells, FRET can be measured by comparing the emission in the donor and acceptor channel at donor excitation and the acceptor fluorescence upon acceptor excitation (ratiometric method). Alternatively, the fluorescence lifetime of
the donor can be measured (Fluorescence Lifetime Imaging Microscopy, FLIM) or the acceptor can be selectively photobleached and the increase of the donor fluorescence can be measured (Acceptor
photobleaching). For cell measurements with genetically encoded fluorescent proteins, the FRET pair CFP and YFP has become popular.
Further Readings:
http://micro.magnet.fsu.edu/primer/techniques/fluorescence/fret/fretintro.html
|
|
2-Photon Microscopy
Principle:
Instead of exciting the fluorophore with the wavelenght of maximum absorption one can also use the doubled wavelength and excite with two photons at a time. This excitation has
several advantages. First, longer wavelength light usually is less photo-toxic, thus the cell damage is minimized. Second, the penetration depth of longer wavelenght light is higher than shorter wavelength light. So imaging up to 1 mm in z-dimension is possible. Last, the z-focus is much smaller with 2-Photon microscopy compared to single photon excitation. Since excitation takes place only where high intensities of photons are achieved, the method provides intrinsically confocal excitation at the focal plane.
Further Readings:
http://micro.magnet.fsu.edu/primer/techniques/fluorescence/multiphoton/multiphotonintro.html
|
 |
|
|
| |
|
|
 |
Single Molecule Imaging
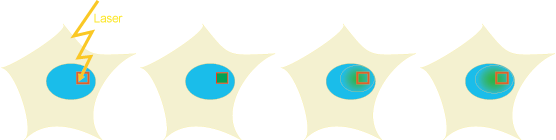
Principle:
Using high sensitivity detectors and fast aquisition sequences it is possible to image single fluorescent molecules. From image sequences mobility parameters and single molecule tracks can be extracted. Properties of single molecules can be studied instead of averaged populations. Also region specific information can be extracted, such as diffusion in different parts of the cell. Importantly, different mobility can be directly determined and a very high spatial and temporal resolution
References:
Grünwald, D., Martin, R.M., Buschmann, V., Bazett-Jones, D.P., Leonhardt, H., Kubitscheck, U., and Cardoso, M.C. (2008)
|
|
|
|
|
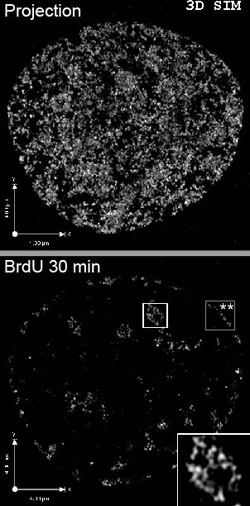
Super-Resolution Microscopy
Principle:
Recent improvements have allowed to go beyond the Abbe diffraction barrier in light microscopy and enable optical resolutions around 100 nm. Several principles are available. 4-Pi microscopy, STED-Microscopy (stimulated emission depletion), GSD Ground State Depletion, and (S)SIM [(saturated) structured illumination microscopy]. Precise location on the single molecule level can be achieved by PALM (photoactivated localization microscopy) or STORM
(stochastic optical reconstruction microscopy).
|
|
 |
References:
Schermelleh, L., Carlton, P.M., Haase, S., Shao, L., Winoto, L., Kner, P., Burke, B., Cardoso, M.C., Agard, D.A.,
Gustafsson, M.G., Leonhardt, H., and Sedat, J.W. (2008)
Baddeley, D., Chagin, V.O., Schermelleh, L., Martin, S., Pombo, A., Carlton, P.M., Gahl, A., Domaing, P., Birk, U., Leonhardt, H., Cremer, C., and Cardoso, M.C. (2010)
Schermelleh, L., Heintzmann, R., and Leonhardt, H. (2010)
Natale, F., Rapp, A., Yu, W., Maiser, A., Harz, H., Scholl, A., Grulich, S., Anton, T., Horl, D., Chen, W., Durante, M., Taucher-Scholz, G., Leonhardt, H. andCardoso, M.C. (2017)
Further Readings:
http://zeiss-campus.magnet.fsu.edu/articles/superresolution/index.html
https://www.microscopyu.com/techniques/super-resolution
|
|
|
|
Light Sheet Microscopy
Principle:
Light sheet microscopy (also known as LSFM - Light Sheet Fluorescence Microscopy or SPIM - Selective Plane Illumination Microscopy) is a modern method of fluorescence microscopy that makes it possible to examine living and thick biological samples in three dimensions and with high temporal resolution - with minimal damage to the sample.
Instead of illuminating the entire sample as with conventional microscopes, a thin light disc (a focused laser beam) is shone into the sample perpendicular to the direction of observation in light disc microscopy. Only the optical plane that is captured by this light disc is excited and observed.
Advantages
Minimised phototoxic effects (less light stress for living samples)
Fast acquisition of 3D data through sequential scanning of many layers
High resolution and contrast in the observed layers
References:
Stelzer, E.H.K., Strobl, F., Chang, BJ. et al. Light sheet fluorescence microscopy. Nat Rev Methods Primers 1, 73 (2021).
https://doi.org/10.1038/s43586-021-00069-4
Further Readings:
https://www.miltenyibiotec.com/DE-en/support/macs-handbook/macs-technologies/light-sheet-microscopy.html
https://www.microscopyu.com/techniques/light-sheet/light-sheet-fluorescence-microscopy
|
|
AIRySCAN Super-Resolution optical Microscopy
Principle:
Airyscan super-resolution microscopy is a high-resolution microscopy technique based on confocal laser scanning microscopy (CLSM), but with significantly improved resolution, sensitivity and signal-to-noise ratio. It was developed by Zeiss and is a super-resolution technique that overcomes several disadvantages of classic oprical super-resolution methods.
In a classic confocal microscope, a pinhole is used to block out light outside the focus. However, a lot of useful signal is lost in the process.
Airyscan technology replaces the conventional pinhole with a detector array with 32 sensitive detectors that can capture the entire Airy disc (the diffraction pattern of a light spot). By reconstructing and analysing this Airy disc with a special algorithm, an image is obtained with:
- Higher resolution (up to 120 nm lateral, approx. 350 nm axial)
- Better signal-to-noise ratio
- More light yield (more sensitive than classic confocal)
References:
Huff, J. The Airyscan detector from ZEISS: confocal imaging with improved signal-to-noise ratio and super-resolution. Nat Methods 12, i–ii (2015). https://doi.org/10.1038/nmeth.f.388
Korobchevskaya, K.; Lagerholm, B.C.; Colin-York, H.; Fritzsche, M. Exploring the Potential of Airyscan Microscopy for Live Cell Imaging. Photonics 2017, 4, 41.
https://doi.org/10.3390/photonics4030041
Further Readings:
https://pages.zeiss.com/rs/896-XMS-794/images/ZEISS-Microscopy_The-Basic-Principle-of-Airyscanning.pdf
|
|
| |
Fluorophores
Using synthetic or covalently linked fluorescent dyes, or in vivo-labeling of cellular structures can be achieved. The most important features for fluorescent dyes are: its absorption and emission maximum; its molecular brightness determined by the extinction coefficient and the quantum yield of the dye. Furthermore, photostability, hydrophobicity and quenching behaviour are important parameters depending on the application.
Providers of Fluorophores:
ATTO-Tec
Denovo Biolabels
Dyomics
GE Healthcare
Invitrogen
For in-vivo labeling, many genetically encoded fluorescent proteins offer unique possibilities for selective staining of structures in living cells, which allows direct
observation of protein dynamics in vivo.
Providers of Fluorescent Proteins:
Clontech
Evrogen
Invitrogen
Fluorescent Proteins
For multicolor applications, the right filter- and excitation settings are crucial. Correct filter settings can be checked at filter manufacturers homepages.
Providers of Optical Filters:
AHF Analysentechnik
Chroma
Omega Optical
Semrock
Filters and dyes - Zeiss |
|
|
|

